

Drug Recommendation Based on Individual Specific Biomarkers
-
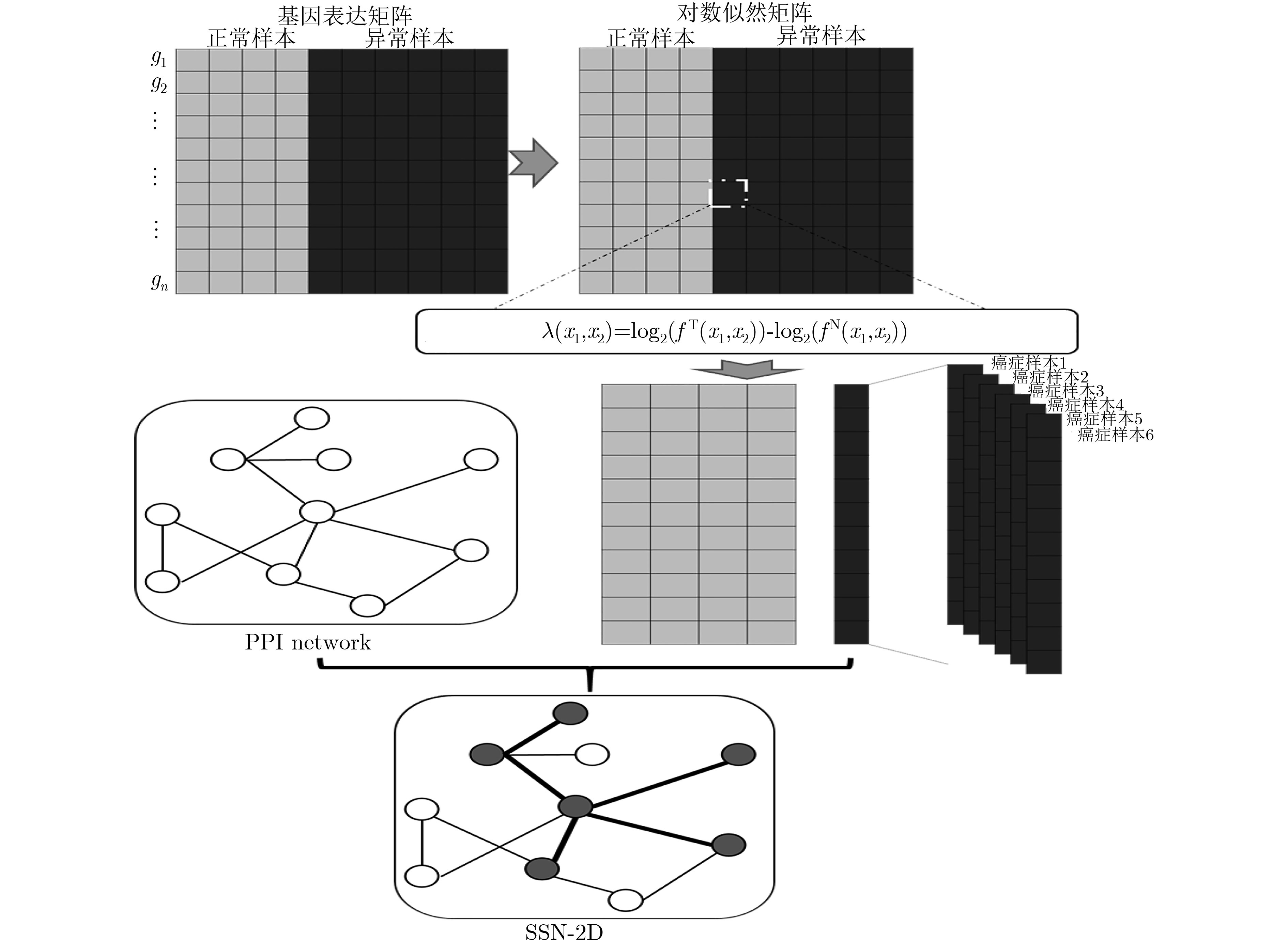
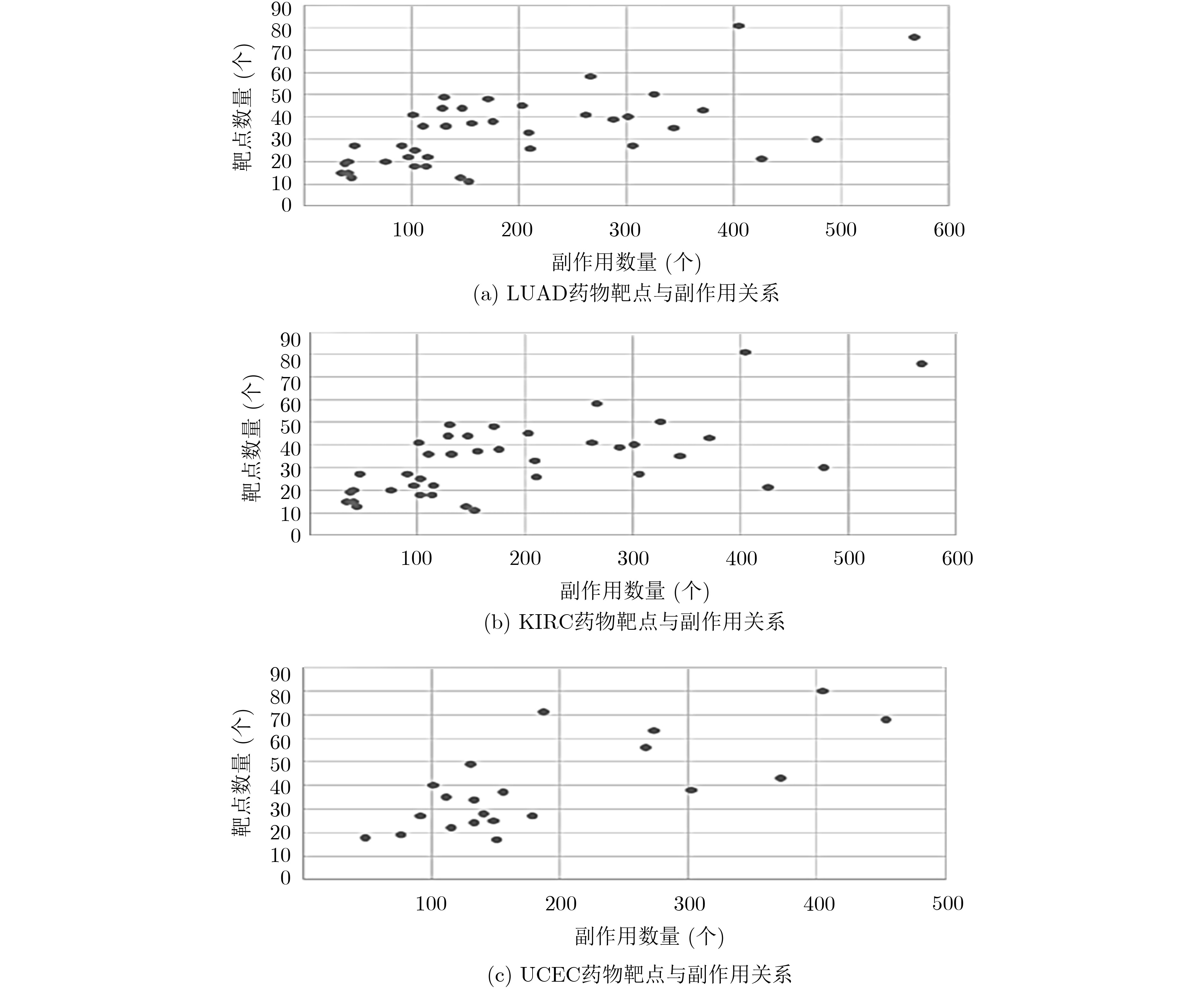
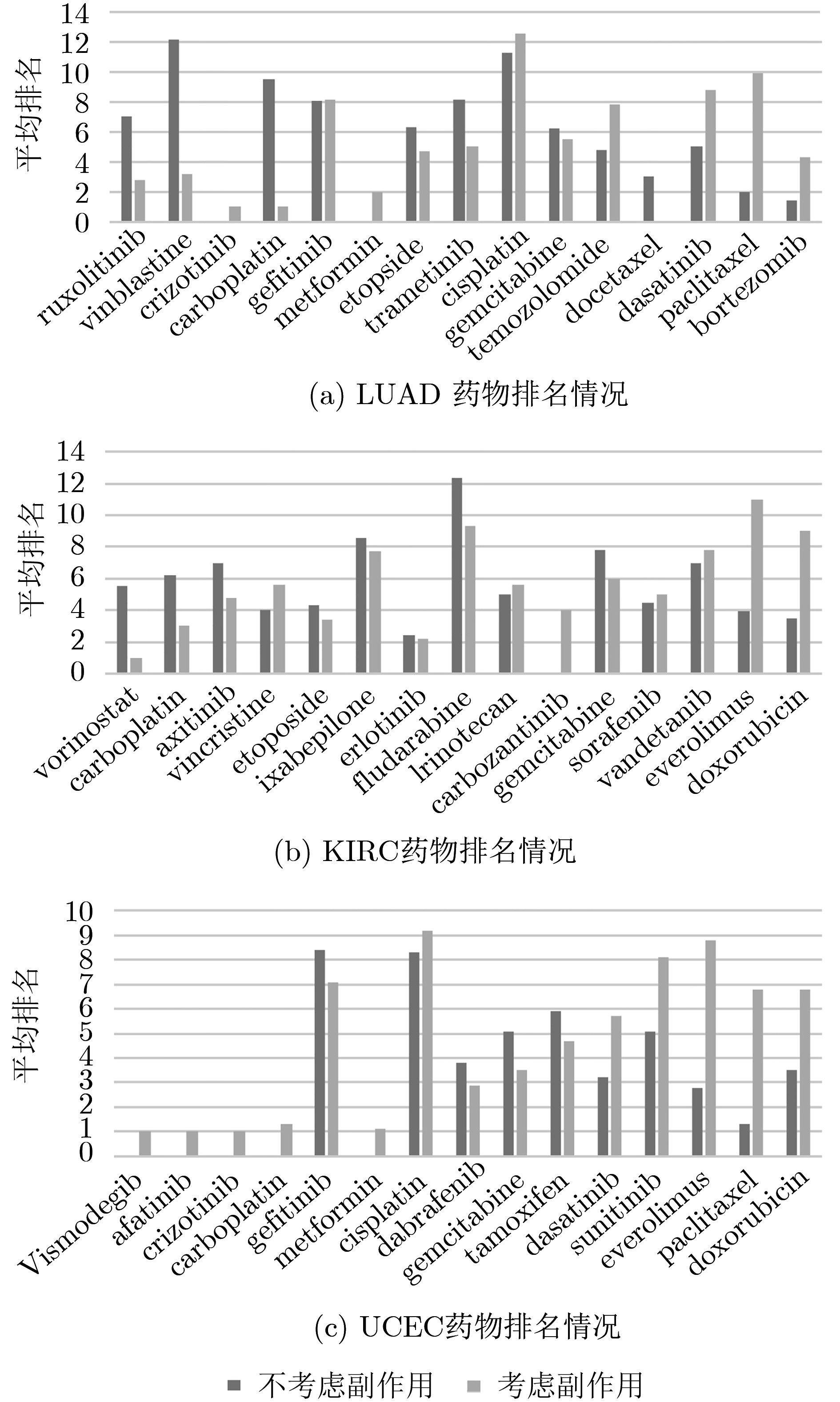
摘要: 基于个性化标志物的药物推荐研究,有助于实现个性化用药及推动精准医疗的发展。该文利用基因表达谱数据及蛋白质网络信息,基于基因2维高斯分布方法筛选出个性化网络标志物。进而综合考虑靶基因的重要性和药物的副作用,提出了一种计算药物对个性化标志物影响权重的方法。将该方法应用于肺腺癌、肾透明细胞癌和子宫内膜癌数据集,通过启发式搜索方法,得到每个疾病样本重要药物推荐列表。结果表明,推荐的药物列表在同种癌症不同样本中既存在一致性,也表现出很大的差异性,如药物种类及药物排序差异,这说明个性化药物在疾病治疗中的重要性及必要性。通过从药物数据库中搜索药物组合对疾病治疗的影响作用表明,该文方法筛选得到的许多药物组合对具体疾病治疗具有积极影响,这进一步证明该文基于个性化网络标志物的药物推荐方法的准确性。该文的研究将有效促进精准化医疗的发展。Abstract: Drug recommendation research based on personalized markers can help to achieve personalized medicine and promote the development of precision medicine. In this paper, a method for calculating the weight of drugs on personalized markers is proposed, which first uses gene expression profile data and protein network information to filter out personalized network markers based on gene two-dimensional Gaussian distribution and then uses the importance degree of genes and the drugs side effect data to calculate the weight of drugs. This method is applied to lung adenocarcinoma, kidney renal clear cell carcinoma and uterine corpus endometrial carcinoma. Through the iterative process, a list of important drug recommendations for each disease sample is got. The results show that there are some differences in the recommended drug list and the ordering importance of drugs in different cases of the same kind of cancer, which indicates the importance and necessity of personalized drugs in the treatment of diseases. By querying the relationship between drugs from the drug database, many of the drug combinations screened by this method have a positive effect on the treatment of specific diseases, which further proves the accuracy of the drug recommendation methods based on personalized network markers. This study will effectively promote the development of precision medicine.
-
Key words:
- Precision medicine /
- Personalized biomarkers /
- Network biomarkers /
- Drug recommendation
-
表 2 启发式搜索的迭代过程
个性化药物推荐算法 输入:物集合$D = \{ d_1,d_2, ··· ,{d_n}\} $; 个性化标志物集合$T = \{ t_1,t_2, ··· ,t_m\} $; 输出:个性化药物推荐列表 (Personalized Drug, PD); (1) Initialization: Set $k = 1$; (2) DO (3) for $i = 1,2, ··· ,n$ (4) Compute $S\left( {{d_i}} \right)$; (5) EndFor (6) If S(di) is the maximum among all drugs in $D$ then (7) ${\rm{PD}}\left( k \right) = {d_i}$; (8) $k = k + 1$; (9) EndIf (10) Update $D$: Delete di from $D$; (11) Update $T$: Delete all targets of ${d_i}$ from $T$; (12) WHILE Max(targets number of each drug in $D$)>=6  下载: 导出CSV
下载: 导出CSV
-
SIEGEL R L, MILLER K D, and JEMAL A. Cancer statistics, 2016[J]. CA: A Cancer Journal for Clinicians, 2016, 66(1): 7–30. doi: 10.3322/caac.21332 TORRE L A, BRAY F, SIEGEL R L, et al. Global cancer statistics, 2012[J]. CA: A Cancer Journal for Clinicians, 2015, 65(2): 87–108. doi: 10.3322/caac.21262 WANG Hongwei, SUN Qiang, ZHAO Wenyuan, et al. Individual-level analysis of differential expression of genes and pathways for personalized medicine[J]. Bioinformatics, 2015, 31(1): 62–68. doi: 10.1093/bioinformatics/btu522 LIU Xiaoping, WANG Yuetong, JI Hongbin, et al. Personalized characterization of diseases using sample-specific networks[J]. Nucleic Acids Research, 2016, 44(22): e164. doi: 10.1093/nar/gkw772 ZHANG Wanwei, ZENG Tao, and CHEN Luonan. EdgeMarker: Identifying differentially correlated molecule pairs as edge-biomarkers[J]. Journal of Theoretical Biology, 2014, 362: 35–43. doi: 10.1016/j.jtbi.2014.05.041 YU Xiangtian, ZHANG Jingsong, SUN Shaoyan, et al. Individual-specific edge-network analysis for disease prediction[J]. Nucleic Acids Research, 2017, 45(20): e170. doi: 10.1093/nar/gkx787 PERLMAN L, GOTTLIEB A, ATIAS N, et al. Combining drug and gene similarity measures for drug-target elucidation[J]. Journal of Computational Biology, 2011, 18(2): 133–145. doi: 10.1089/cmb.2010.0213 WANG Wenhui, YANG Sen, and LI Jing. Drug Target Predictions Based on Heterogeneous Graph Inference[M]. ALTMAN R B, DUNKER A K, HUNTER L, et al. Biocomputing 2013. Hawaii, USA: World Scientific, 2013: 53-64 doi: 10.1142/9789814447973_0006. ZONG Nansu, KIM H, NGO V, et al. Deep mining heterogeneous networks of biomedical linked data to predict novel drug-target associations[J]. Bioinformatics, 2017, 33(15): 2337–2344. doi: 10.1093/bioinformatics/btx160 SU Junjie, YOON B J, and DOUGHERTY E R. Accurate and reliable cancer classification based on probabilistic inference of pathway activity[J]. PLoS One, 2009, 4(12): e8161. doi: 10.1371/journal.pone.0008161 TOMCZAK K, CZERWIŃSKA P, and WIZNEROWICZ M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge[J]. Contemporary Oncology (Poznan, Poland) , 2015, 19(1A): A68–A77. doi: 10.5114/wo.2014.47136 SZKLARCZYK D, MORRIS J H, COOK H, et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible[J]. Nucleic Acids Research, 2017, 45(D1): D362–D368. doi: 10.1093/nar/gkw937 WISHART D S, FEUNANG Y D, GUO A C, et al. DrugBank 5.0: A major update to the DrugBank database for 2018[J]. Nucleic Acids Research, 2018, 46(D1): D1074–D1082. doi: 10.1093/nar/gkx1037 GRIFFITH M, GRIFFITH O L, COFFMAN A C, et al. DGIdb: Mining the druggable genome[J]. Nature Methods, 2013, 10(12): 1209–1210. doi: 10.1038/nmeth.2689 李玉博, 陈邈. 几乎完备高斯整数序列构造法[J]. 电子与信息学报, 2018, 40(7): 1752–1758. doi: 10.11999/JEIT170844LI Yubo and CHEN Miao. Construction of nearly perfect gaussian integer sequences[J]. Journal of Electronics &Information Technology, 2018, 40(7): 1752–1758. doi: 10.11999/JEIT170844 陈曦, 张坤. 一种基于树增强朴素贝叶斯的分类器学习方法[J]. 电子与信息学报, 2019, 41(8): 2001–2008. doi: 10.11999/JEIT180886CHEN Xi and ZHANG Kun. A classifier learning method based on tree-augmented naïve bayes[J]. Journal of Electronics &Information Technology, 2019, 41(8): 2001–2008. doi: 10.11999/JEIT180886 FANUCCHI M P, FOSSELLA F V, BELT R, et al. Randomized phase ii study of bortezomib alone and bortezomib in combination with docetaxel in previously treated advanced non-small-cell lung cancer[J]. Journal of Clinical Oncology, 2006, 24(31): 5025–5033. doi: 10.1200/JCO.2006.06.1853 DAVIES A M, RUEL C, LARA P N, et al. The proteasome inhibitor bortezomib in combination with gemcitabine and carboplatin in advanced non-small cell lung cancer: A california cancer consortium phase I study[J]. Journal of Thoracic Oncology, 2008, 3(1): 68–74. doi: 10.1097/JTO.0b013e31815e8b88 DAVIES A M, CHANSKY K, LARA Jr P N, et al. Bortezomib plus gemcitabine/carboplatin as first-line treatment of advanced non-small cell lung cancer: A phase II southwest oncology group study (S0339)[J]. Journal of Thoracic Oncology, 2009, 4(1): 87–92. doi: 10.1097/JTO.0b013e3181915052 KELLY K, CROWLEY J, BUNN PA, et al. Randomized phase III trial of paclitaxel plus carboplatin versus vinorelbine plus cisplatin in the treatment of patients with advanced non-small-cell lung cancer: A southwest oncology group trial[J]. Journal of Clinical Oncology, 2001, 19(13): 3210–3218. doi: 10.1200/JCO.2001.19.13.3210 SANDLER A, GRAY R, PERRY M C, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer[J]. New England Journal of Medicine, 2006, 355(24): 2542–2550. doi: 10.1056/NEJMoa061884 MCNAMARA M, SWEENEY C, ANTONARAKIS E S, et al. The evolving landscape of metastatic hormone-sensitive prostate cancer: A critical review of the evidence for adding docetaxel or abiraterone to androgen deprivation[J]. Prostate Cancer and Prostatic Diseases, 2018, 21(3): 306–318. doi: 10.1038/s41391-017-0014-9 ROBINET G, BARLESI F, FOURNEL P, et al. Second-line therapy with gefitinib in combination with docetaxel for advanced non-small cell lung cancer: A phase II randomized study[J]. Targeted Oncology, 2007, 2(2): 63–71. doi: 10.1007/s11523-007-0042-9 ANGELOPOULOU A, KOLOKITHAS-NTOUKAS A, FYTAS C, et al. Folic acid-functionalized, condensed magnetic nanoparticles for targeted delivery of doxorubicin to tumor cancer cells overexpressing the folate receptor[J]. ACS Omega, 2019, 4(26): 22214–22227. doi: 10.1021/acsomega.9b03594 TEDESCO-SILVA H, PASCUAL J, VIKLICKY O, et al. Safety of everolimus with reduced calcineurin inhibitor exposure in de novo kidney transplants: An analysis from the randomized TRANSFORM study[J]. Transplantation, 2019, 103(9): 1953–1963. doi: 10.1097/TP.0000000000002626 -
 下载:
下载:



图(5) / 表(2)
计量
- 文章访问数: 3309
- HTML全文浏览量: 1457
- PDF下载量: 258
- 被引次数: 0
